Więcej o Chorobie Sanfilippo
Choroba Sanfilippo inaczej MPS III czyli mukopolisacharydoza typu III nazwa pochodzi od nazwiska lekarza z USA – dr Sylvestra Sanfilippo, który opisał ta chorobę w 1963 roku. Jak dotąd, nie ma lekarstwa na chorobę Sanfilippo ale naukowcy poszukują sposobu leczenia tego zaburzenia.
Co to jest zespół Sanfilippo?
Mukopolisacharydy są to długie łańcuchy cząsteczek cukru wykorzystywane w budowie kości, chrząstki, skóry, ścięgna i wielu innych tkanek w organizmie. Od pewnego czasu słowo Mukopolisacharydy zastępuje się nazwą glikozaminoglikany lub GAG. W trakcie całego życia, istnieje ciągły proces budowania nowych Mukopolisacharydów i rozbicia starych. Ten nieustanny proces jest konieczny do utrzymania zdrowego ciała. Ten rozkład mukopolisacharydów wymaga specjalnych narzędzi zwanych biochemicznymi enzymami. Osoby z MPS III brakuje jednego z czterech specyficznych enzymów, które są niezbędne w rozbiciu jednego z GAG o nazwie siarczan heparyny. W podziale heparyny niepełny, nierozłozony siarczan nadal przechowywany jest wewnątrz komórek w organizmie i tworzy przyczynę postępującego uszkodzenia. GAG nie jest toksyczny, ale jego ilość oraz wpływ przechowywania go w organizmie, a zwłaszcza w mózgu prowadzi do wielu problemów fizycznych, takich jak opóźniony rozwój i nadpobudliwość, zaburzenia snu, utrata mowy, demencja i prowadzi do śmierci przed ukończeniem pełnoletności . Małe dzieci zazwyczaj nie wykazują objawów choroby, jednak w miarę upływyu czasu GAG gromadzi się i objawy pojawiają się zwykle w wieku około 2 – 6 lat.
Różne formy choroby
Istnieją cztery różne braki enzymów, które powodują chorobę Sanfilippo i są one opisane jako typ A, B, C lub D. Enzymy te to N-sulfatazy heparyny (typ A), alfa-N-acetylglucosaminidase (typ B), acetylo-CoA-glucosaminide acetylotransferazę (typu C) i N-acetylglucosamine-6-sulfatazy (typ D). Istnieje niewiele klinicznej różnicy między tymi czterama typami Sanfilippo Wszystkie cztery typy kumulują w organiźmie ten sam GAG – siarczan heparyny. To przedewszystkim siarczan heparyny odkłada się w ośrodkowym układzie nerwowym i to nagromadzenie w mózgu jest przyczyną wielu problemów, które dotykają osoby ze wszystkimi typami Sanfilippo.
| Typ MPS III | Brakujący enzym | Lokalizacja w genie |
|---|---|---|
| MPS III typ A | heparan N-sulfatazy | 17q25.3 |
| MPS III typ B | N-acetylo-alfa-D-glucosaminidasa | 17q21 |
| MPS III typ C | acetylo-CoA-glucosaminide acetyltransferase | 8p11-P13 |
| MPS III typ D | N-acetylglucosamine-G-siarczan sulfatazy |
12q14 |
Jak częste jest MPS III?
Częstość występowania MPS III (wszystkie cztery rodzaje łącznie) szacuje się na 1 na 70.000 urodzeń. Typ A jest najczęstszy i najczęściej spotykany jest w Europie północno-zachodniej, typ B w Europie Południowo-Wschodniej, typy C i D są bardzo rzadkie. Sanfilippo zaliczane jest do rzadkich chorób .W Polsce (dane z 2000 roku) częstość występowania tej choroby szacuje się na 1 na 66.000 żywych urodzeń. W poszczególnych typach szacuje się częstość wystepowania:
| MPS IIIA 1 na 114.000 żywych urodzeń |
| MPS IIIB 1 na 211.000 żywych urodzeń |
| MPS IIIC i MPS IIID są o wiele rzadsze. |
Jak dziedziczony jest zespół Sanfilippo?
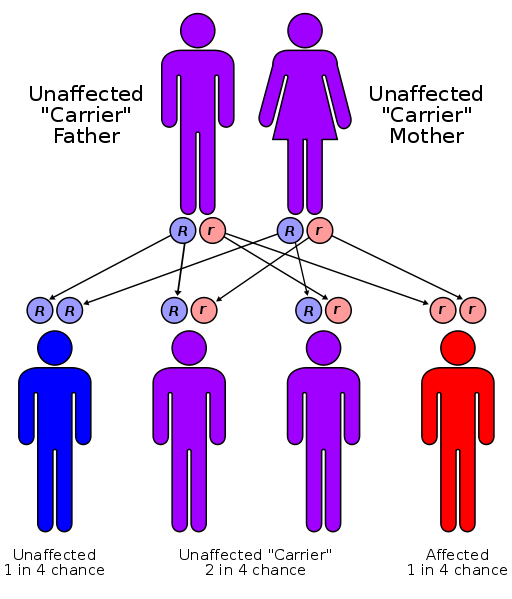
Większość MPS, w tym choroba Sanfilippo ma autosomalny recesywny sposób dziedziczenia. Oznacza to, że zaburzenie występuje tylko wtedy, gdy oboje rodzice noszą to zaburzenie genu. Gdy oboje rodzice mają takie zaburzenia genów, jest prawdopodobieństwo ¼ że będą posiadać dziecko chore na Sanfilippo lub inaczej 25% na urodzenie chorego dziecka w każdej ciąży. Jest także prawdopodobieństwo 2/3 że dziecko będzie nosicielem choroby tak jak jego rodzice i 1/3 szansy że dziecko będzie zupełnie zdrowe. Większość rodzin u których urodziło się dziecko z MPSIII nie miało wcześniej problemów genetycznych w przeprowadzonym wywiadzie rodzinnym . Enzymatyczna wada wystepująca we wszystkich typach zespółu Sanfilippo jest dziedziczona w sposób autosomalny recesywny ( dotyczy również innych zaburzeń MPS, za wyjątkiem MPS II (tj. zespołem Huntera), które jest połączone X-recesywnym sposobem dziedziczenia). Mutacja genu znajduje się w autosomie, a nie w chromosomach płci ,w związku z tym dotyka zarówno chłopców jak i dziewczynki.
Jak wygląda postęp choroby?
Zespół Sanfilippo dotyka dzieci w różny sposób i rozwija w bardzo różnym tempie. Dzieci z zespołem Sanfilippo rodzą się bez objawów choroby, zazwyczaj zupełnie zdrowe i zazwyczaj ich rozwój przebiega normalnie do pierwszych 2 lat życia. Głównym objawem tej choroby jest regresja rozwoju psychoruchowego, który występuje pomiędzy 2 i 6 rokiem życia dziecka . Początkowe objawy kliniczne występują w postaci zmian w zachowaniu. Dzieci mogą mieć zaburzenia zachowania , problemy emocjonalne, opóźnienie psychomotoryczne, nadpobudliwość z cechami autyzmu , zaburzenia behawioralne. Podczas wczesnych lat szkolnych objawy zazwyczaj się nasilają i dziecko ma trudności z koncentracją i pamięcią . Spowalnia się rozwój umysłowy , a następnie dochodzi do stopniowego pogorszenia chodu i artykulacji mowy. Postęp choroby jest stopniowy i można mówic o trzech etapach choroby. Dziecko w wieku przedszkolnym jest bardzo absorbujące i frustrujące dla rodziców. Dziecko zaczyna „odstawać” od swoich rówieśników ,często rodzice są obwiniani za jego złe zachowanie i problemy z nauką. Często diagnoza jest stawiana bardzo późno, ponieważ niektóre dzieci nie mają żadnych zmian w wyglądzie, a ich objawy w zachowaniu są przypisywane osobowości i złemu wychowaniu. Lekarz musi być bardzo spostrzegawczy, aby rozpoznać objawy i powiazac je z chorobą Sanfilippo.
Zdarza się, że w rodzinie jest dwoje dzieci dotknietych przez chorobę.Zazwyczaj gdy dowiadują się o chorobie starszego dziecka na świecie jest już drugie dziecko ,które okazuje się być także chore na Sanfilippo. Drugi etap choroby charakteryzuje się niezwykłą aktywnością, ruchliwością i zachowaniem nie akceptowanym przez otoczenie.. Niektóre dzieci bardzo mało śpią w nocy.Przeważnie wkładają wszystko do buzi, lubią żuć ręce, ubrania lub cokolwiek, co mogą złapać do ręki. Niestety, język i zrozumienie będzie stopniowo tracony i rodzice mają utrudniony kontakt z dzieckiem. Ale niektóre dzieci potrafią znaleźć inne sposoby komunikowania się. Niektóre dzieci nigdy nie nauczą się korzystania z toalety,a pozostałe utracą tą umiejętnoośc. W trzeciej fazie choroby, dzieci z MPS III zaczynają spowalniać. Stoją niepewnie na nogach,często tracą równowagę,maja trudnośc z poruszaniem się. Ostatecznie tracą one zdolność chodzenia. Życie dziecka zamyka się w czterech ścianach pokoju.Rodzice będą potrzebować pomocy w męczącej fizycznie opiece nad dzieckiem lub nieuruchomionym nastolatkiem.
Częste dolegliwości:
- Dzieci mają częste przeziębienia, katar i chroniczne infekcje ucha powodujace utratę słuchu i przerost trzeciego migdałka
- Więcej agresywnych zachowań było zgłaszane u dziewczynek chorych na Sanfilippo.
- U dzieci występują powtarzające się lub przewlekłe biegunki- za przyczynę tego zaburzenia jelit uważa się akumulację glikozaminoglikanów w neuronach splotu nerwowego błony mięśniowej jelita
- Napady padaczkowe mogą wystąpić w późniejszym okresie choroby, ale zwykle są dobrze kontrolowane za pomocą leków.
- W Sanfilippo tak jak w innych Mukopolisacharydozach (np. zespół Hurler’a, zespół Scheie, zespół Sanfilippo) może powodować retinopatię barwnikową taką jak Retinitis Pigmentosa (RP).Pacjenci mogą okazywać tródność z poruszaniem się w nocy lub w ciemnych miejscach,np. problemy z chodzeniem w źle oświetlonym pomieszczeniu (np. kino). Mogą mieć także problemy z poruszaniem się o zmierzchu lub we mgle lub potrzebować więcej czasu na dostosowanie z jasnego do ciemnego
- Pacjentów z mukopolisacharydozą (MPS) mają większe ryzyko powikłań podczas znieczulenia ogólnego ,należy o tym pamiętać przy zabiegach chirurgicznych . Oprócz ogólnych infiltracji i zgrubienia tkanek miękkich, część ustna gardła może być zasłonięta przez duży język z powiększonym migdałem. Również kruchość błony śluzowej jamy ustnej i gardła powoduje, że łatwo w tych strukturach o krwawienie i obrzęki. Szyja jest zwykle krótka i nieruchoma a kręgosłup szyjny może mieć ograniczony zakres ruchu. Budowa anatomiczna i wrażliwość układu oddechowego powodują trudności w intubacji lub może dochodzić do skurczu oskrzeli po intubacji.
- Zmiany w kościach są łagodne, ale u części pacjentów może rozwijać się dysplazja bioder i zmiany w miednicy ( które można zobaczyć na radiografii ), łagodne dysostosis multipleks (charakterystyczna nieprawidłowość szkieletowa w MPS ), może to powodować ból i sztywność stawów oraz często diagnozę choroby Perthe.
Badania
We wszystkich typach MPS III występuje zwiększone wydalanie siarczanu heperanu z moczem. Diagnoza MPSIII opiera się właśnie na oznaczeniu poziomu HS w moczu. Do pomiaru stężenia glikozaminoglikanów (GAG) w moczu. Dla wszystkich rodzajów Sanfilippo może być zbadana aktywność enzymatyczna przez oznaczenie w hodowanych fibroblastach skóry i leukocytów krwi obwodowej
Zalecane badania:
- neuroobrazowanie (tomografia komputerowa) do diagnostyki wodogłowia i określenia zmian w strukturze mózgu
- echokardiografia
- USG serca
- badanie jamy brzusznej, takie jak ultrasonografia lub tomografia komputerowa do oceny powiększenia narządów
- elektroencefalografia – zapis czynności elektrycznej mózgu (EEG) do diagnozowania czynności napadowych
- badanie audiologiczne w celu rozpoznania pacjentów z uszkodzeniem słuchu
- polisomnografia dla tych pacjentów, którzy wykazują kliniczne objawy obturacyjnego bezdechu podczas snu (OBPS).
Na podstawie:
https://www.emedicine.com/ped/topic2040.html
https://www.orpha.net/actor/Orphanews/2007/doc/SANFILIPPO_White_Paper.pdf