O badaniach
Za kilka lat choroba taka jak Sanfilippo może nie być już nieuleczalna
Nad nowymi metodami leczenia pracuje Profesor Grzegorz Węgrzyn. Kilka lat temu opracował pierwszą na świecie metodę pozwalającą na zahamowanie choroby Sanfilippo.
Substancja pozyskiwana z soi – genisteina okazała się skutecznie hamować drogę przekazywania sygnałów wewnątrz komórki, prowadząc do obniżenia poziomu syntezy glikozoaminoglikanów – substancji, których nadmiar w komórkach cierpiących na mukopolisacharydozy, jest przyczyną objawów chorobowych. Tym samym genisteina może spowalniać rozwój choroby.
W Polsce przeprowadzono już pilotażowe badania kliniczne. Niebawem w Holandii rozpocznie się II/III faza badań klinicznych z zastosowaniem podwójnie ślepej próby z placebo. Każdy lek przed wprowadzeniem na rynek musi przejść szereg badań, a każdy z etapów prób klinicznych musi być prowadzony zgodnie z obowiązującym prawem i z wymogami międzynarodowymi. Nowy preparat musi być dokładnie zbadany pod względem skuteczności, optymalnych dawek i skutków ubocznych. Dlatego badania takie są bardzo kosztowne – wymagają wykorzystania wielu specjalistycznych urządzeń i pracy wielu ludzi.
Zespół naukowców pod kierownictwem Profesora Grzegorza Węgrzyna pracuje nad wykorzystaniem do leczenia Sanfilippo pochodnych genisteiny, które charakteryzowałyby się większą skutecznością i efektywnością m.in. w pokonywaniu bariery krew-mózg. Terapia genisteiną nie przynosi efektu całkowitego wyleczenia, dlatego wciąż poszukuje się nowych opcji terapeutycznych.
Zobacz wywiad z Prof.dr hab.Grzegorzem Węgrzynem dowiesz się więcej o chorobie Sanfilippo i badaniach nad lekiem.
Aktualności naukowe
Średnio 1 na 10 Amerykanów i 250 mln ludzi na świecie dotkniętych jest chorobą rzadką. Choroby rzadkie, często również określane…
Czytaj więcej
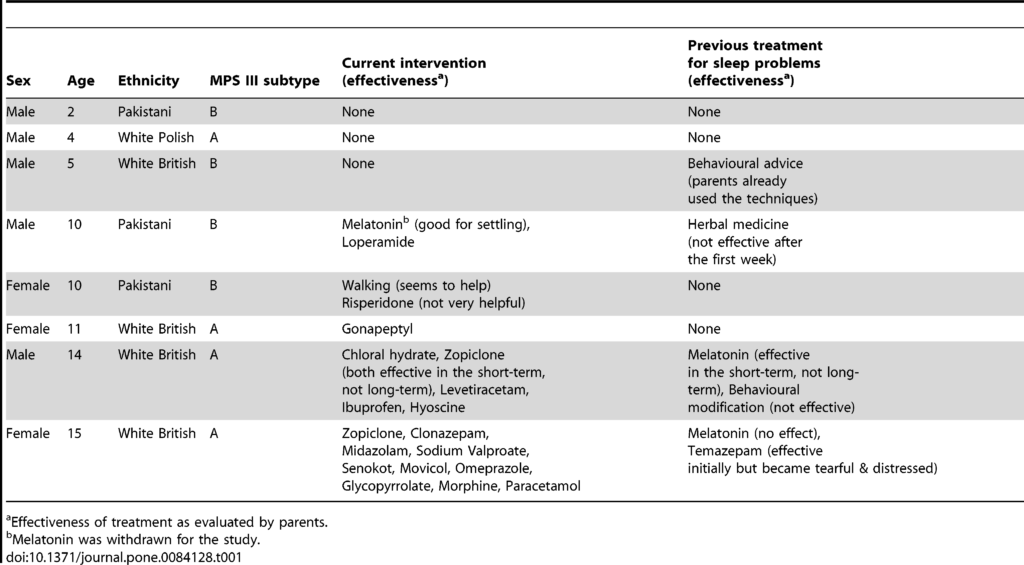
Głównym objawem Mukopolisacharydozy typu III są zaburzenia snu, obecnie brak jest obiektywnych,wiarygodnych, udokumentowanych badań opisujących ilość oraz jakość snu czy…
Czytaj więcejOpracowana przez włoskich lekarzy terapia genowa umożliwiła pokonanie nieuleczalnych dotąd chorób. Dzięki wykorzystaniu zmodyfikowanego wirusa HIV specjalistom z Istituto San…
Czytaj więcej
Stowarzyszenie chorych z MPS w Wielkiej Brytanii przyznało pierwszy grant w wysokości £ 160.000 Uniwersytetowi w Manchesterze na sfinansowanie "Fazy…
Czytaj więcejFirma ArmaGen Technologies Inc. poinformowała w komunikacie prasowym o pozyskaniu finansowania w kwocie 17.000.000 dolarów, które zostanie wykorzystane na opracowywanie…
Czytaj więcej

GlaxoSmithKline nawiązało kontakt z kanadyjską firmą Angiochem aby odkryć, rozwinąć oraz sprzedawać terapie dla lizosomalnych chorób spichrzeniowych, takich jak choroba…
Czytaj więcejW dniach 8-10 lutego 2012 w San Diego USA odbyła się Międzynarodowa Konferencja dotycząca badań nad chorobami lizosomalnymi.To co najbardziej…
Czytaj więcej
Trwają obecnie badania w wielu ośrodkach naukowych nad wykorzystaniem genisteiny w leczeniu chorych na Sanfilippo. Dla rodzin i chorych dzieci…
Czytaj więcejPodsumowanie międzynarodowej konferencji poświęconej badaniom w kierunku leczenia mukopolisacharydoz
W dniach 8-10 grudnia 2011 r. w Genewie odbyła się międzynarodowa konferencja poświęcona badaniom dotyczącym leczenia mukopolisacharydoz. Naukowcy i…
Czytaj więcej
CHOROBA SANFILIPPO, MUKOPOLISACHARYDOZA TYPU III, JEST RZADKIM SCHORZENIEM GENETYCZNYM. Powoduje degradację układu nerwowego, wyniszcza i prowadzi do śmierci. Chorzy, których…
Czytaj więcej18 lipca francuska firma farmaceutyczna LYSOGENE poinformowała, że uzyskała zgodę AFSSAPS (Agencji ds. bezpieczeństwa sanitarnego produktów zdrowotnych) oraz komisji bioetycznej…
Czytaj więcej

Amsterdam Molecular Therapeutics (AMT), lider w ludzkiej terapii genowej, ogłosił dzisiaj zawarcie porozumienia z Instytutem Pasteura w Paryżu oraz grupą…
Czytaj więcejTak niewiele trzeba aby pomóc dzieciom chorym na Sanfilippo! Wystarczy że wejdziesz na stronę i zagłosujesz - można głosować raz…
Czytaj więcej

Firma Shire wystosowała list do rodziców dzieci chorych na Sanfilippo w związku z licznymi pytaniami dotyczącymi prób klinicznych z leczeniem…
Czytaj więcejShire Human Genetic Therapies (Shire HGT) rozwija terapię zastępczą enzymem sulfamidowym (ERT)rhHNS przeznaczoną dla pacjentów z MPS IIIA. Enzym rhHNS…
Czytaj więcej

Głównym celem Stowarzyszenia Uratujmy Życie jest zebranie niezbędnych funduszy na badania nad lekiem ratującym życie dzieciom chorym na Sanfilippo. Mocno…
Czytaj więcejAutor: mgr Marcelina Malinowska UG Mukopolisacharydozy (MPS) to grupa rzadkich chorób genetycznych, charakteryzująca się gromadzeniem niekompletnie zdegradowanych glikozaminoglikanów (GAG) w…
Czytaj więcej

PRZEKRÓJ | 25 czerwca 2009 Anna Szulc Osiągnięcia profesora Grzegorza Węgrzyna biologa molekularnego i genetyka, są policzalne. Wystarczy wymienić 200…
Czytaj więcejJest to skrót wyników badań naukowych zakresie terapii genisteiną na myszach z MPSIII - w najbliższym czasie zamieścimy więcej szczegółowych…
Czytaj więcej
Dr Stanislav Karsten, jest pracującym w Los Angeles badaczem w zakresie biomedycyny oraz wiodącym autorem badania. Autor artykułu: Dana Maione.…
Czytaj więcejŹródło: Gazeta Wyborcza Oprac. Alicja Katarzyńska W Gdańsku powstaje lek na nieuleczalną dotąd chorobę genetyczną. Czeka na niego kilka tysięcy…
Czytaj więcej
Autor: Krzysztof Klinkosz, PAP - Nauka w Polsce Zespół polskich naukowców, pod kierunkiem prof. dr hab. Grzegorza Węgrzyna z Uniwersytetu…
Czytaj więcejAutor: dr n. biol. Marta Koton-Czarnecka, Puls Medycyny 14 (137) Naukowcy z Katedry Biologii Molekularnej Uniwersytetu Gdańskiego opracowali pierwszą na…
Czytaj więcej
Źródło: Onet.pl "Rzeczpospolita": Polscy uczeni mają za sobą pomyślny rok. Przeprowadzali pionierskie operacje, badali kosmos, odkrywali starożytne zabytki - czytamy…
Czytaj więcejŹródło: PAP Za odkrycie mechanizmu interferencji RNA, które może mieć zastosowanie w terapii genowej, tegoroczną Nagrodę Nobla w medycynie otrzymali…
Czytaj więcej
Onet.pl/PAP Sztuczne chromosomy, zawierające "terapeutyczny" gen, mogą naprawiać wady genetyczne, będące podłożem poważnych chorób i zaburzeń - wynika z najnowszych…
Czytaj więcejŹródło: Onet.pl/PAP Stosując impulsy elektryczne o odpowiednim napięciu można wprowadzać do żywych komórek fragmenty materiału genetycznego, który następnie może modyfikować…
Czytaj więcej
Autor: Marta Kamieniak / Sprawy Nauki Koncepcja genów kontrolujących inteligencję, osobowość i uzdolnienia niebezpiecznie wciska się w sferę pojęć, za…
Czytaj więcej